Auch wenn der ganze Artikel eher "trocken" klingt, so ist die kritische Beurteilung von Meßwerten extrem wichtig im gesamten technischen und wissenschaftlichen Bereich; denn wie sich zeigt: Jede Messung ist falsch....
Eine normale Analyse z.B. von Pflanzenproben verläuft in der Praxis in drei Schritten:
- Probenvorbereitung: Sammeln von Proben, trocknen, mahlen, aufschließen
- Analysieren
- Auswertung der Analysenergebnisse, Statistik, Labordatenverwaltung
Schon bei der Probenvorbereitung können Fehler auftreten.
Eingrenzen kann man im allgemeinen Fehler, wenn man mehrere Vorgehensweisen (sog. Methoden) unter einander vergleicht, und so Rückschlüsse auf den tatsächlichen Wert zieht. Das ist in der Praxis meist zeit- und kostenaufwendig, so daß es in der Regel nur einmal bei der sog. Methodenentwicklung gemacht wird und dann für mehr oder weniger allgemeingültig erklärt wird. In den Laboratorien werden pro Jahr hunderte solcher Proben bestimmt. Daher ist leicht einzusehen, daß für eine einzelne Probe kaum Zeit bleibt, einen exakten Wert zu ermitteln. Ein Restfehler bleibt daher immer bestehen, schon alleine aus der Tatsache heraus, daß z.B. jede Pflanzenprobe etwas unterschiedlich aufgebaut ist.
Beispiel: Restfeuchte im Pfanzenproben
Beispiel: Beinhaltet die Probe z.B. noch 5% Wasser, so sind die zu analysierenden Mineralstoffe gegenüber der trockenen Probe um 5% "verdünnt" und um diesen Betrag falsch.
Andererseits darf man nicht zu stark erhitzen oder zu lange trocknen, da ein Teil der flüchtigen organischen Bestandteile verdampfen kann und so die Probe scheinbar mit den Mineralstoffen angereichert wird.
Verwendete Wasserbestimmungsmethoden
- Die Karl-Fischer-Methode
- Die Gravimetrische Methode
Zur Wasserbestimmung in Substanzen gehört die Karl-Fischer-Methode zu den genauesten standardisierten Wasserbestimmungsmethoden im Laborbereich. Sie beruht auf der gut untersuchten chemischen Reaktion:
I
2+ SO2+ 2 H2O -> H2SO4+ 2 HI
Das durch Wasser gebildete HI wird mit elektrischen Methoden bestimmt.
Die Probe wird in einem kleinen Becherglas eingewogen, die Einwaage notiert und anschließend eine bestimmte Zeit bei einer bestimmten Temperatur im Trockenschrank, einer Art "Backofen", getrocknet. Danach wird die abgekühlte Probe abermals gewogen und die Massendifferenz berechnet.
Durchführung
Ausgangsprobe : bei ca 30°C drei Tage vorgetrocknete und anschließend gemahlene Buchenblätter.
100,0 mg der Pflanzenprobe wird bei 85°C, 95°C und 105°C im trockenen Stickstoffstrom ausgeheizt und die feuchten Gase der Karl-Fischer-Zelle zugeführt, in der das Wasser selektiv und quantitativ (d.h. vollständig) bestimmt wird.
Als Vergleich werden im Trockenschrank Teile der Probe bei 85°C , 95 °C und 105 °C genau zwei Stunden lang ausgeheizt, im Exsikkator (einem im Labor üblichen topfförmigen Glasgefäß) über Silicagel ® (poröse granulierte Kieselerde als Trocknungsmittel) genau 30 min abgekühlt und die Massendifferenz bestimmt.
Ergebnis
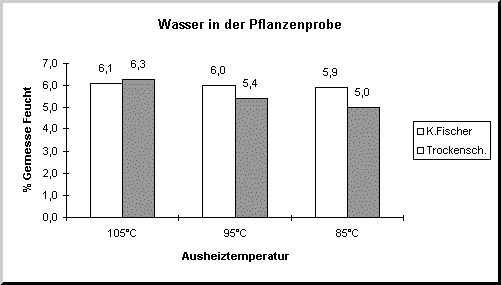
Das Diagramm zeigt deutlich die Differenz zwischen der Karl-Fischer- und der Trockenschrankmethode.
Bei 105°C ergeben beide Methoden ungefähr den gleichen Wert, während bei 85°C zu wenig Wasser im Trockenschrank ausgetrieben wird.
Die Meßbedingungen (wie Temperatur, Zeiten) müssen aber bei allen Vergleichsmessungen immer genau eingehalten werden.
Bei der Spurenanalyse, wo minimalste Mengen z.B. von Schwermetallen oder Pestiziden bestimmt werden sollen, sind Fehler von +/- 20 % und höher keine Seltenheit und aus meßtechnischer Hinsicht verständlich. Bei uns werden Planzenproben in der Regel mit +/- 2-5 % angegeben.
Allgemeine Fehlerbetrachtungen:
Wie angedeutet treten bei jeder Messung grundsätzlich Fehler auf. Diese sind auf die Meßgenauigkeit der verwendeten Geräte, der Meßmethode und der individuellen Sorgfalt des Analytikers zurückzuführen.
Fehlerangaben besagen nicht, das ein Ergebnis falsch sein muß, sondern nur um diesen Betrag falsch sein kann.
Der absolute Fehler wird mit der gleichen Einheit wie der Meßwert z.B. 147 mg +/- 3 mg angegeben.
Der relative Fehler wird dagegen in Prozent z.B. 147 mg +/- 2 % ausgedrückt.
Grobe Fehler z.B. durch falsches Ablesen oder Verrechnung, kann man vermeiden. Durch Wiederholung der Messung (deshalb mindestens eine Doppelbestimmung durchführen) lassen sie sich oft erkennen.
Den systematischen Fehlern kann man dagegen schwer ausweichen, da man ihre Ursachen nicht kennt und auch aus der Messung nicht unbedingt auf sie schließen kann. Wenn z.B. eine falsch eingestellte Waage statt den tatsächlichen 15,52 g nur 15,19 g anzeigt, wird das Ergebnis, auch bei noch so guter Durchführung der Analyse, nie richtig sein.
Zufällige Fehler sind das eigentliche Gebiet der Fehlerrechnung. Diese können nur durch mehrfach ausgeführte Analyse und der Erfahrung des Analytikers erkannt und behoben werden. Es kann es z.B. vorkommen, daß eine eingesandte Holzprobe, die auf ihren Eisengehalt bestimmt werden soll, mit einem Stahlsägeblatt genommen wurde. So kann der Abrieb einer Säge in die Proben gelangen und zu gänzlich falschen Ergebnissen führen. Bei diesem tatsächlich aufgetretenen Ereignis waren ca. 1/3 der abgelieferten Proben für die Eisenbestimmung im Holz unbrauchbar.
In der Praxis wird eine Probe 2-3 mal bestimmt. Die Meßwerte dürfen sich dabei nur unwesentlich unterscheiden , ansonsten muß noch einmal analysiert werden. Treten wieder Unterschiede auf, so liegt es meisten daran, daß die zu untersuchende Probe nicht homogen genug ist.
Literatur
W. Lück: Feuchtigkeit , Oldenbourg Verlag, München, 1964
Analytikum: Methoden der analytischen Chemie und ihre theoretischen Grundlagen, Dt. Vlg. für Grundstoffindustrie, Leipzig, 1979
 Photometrie Photometrie |
Home |
Start |